这是之前的一篇:石墨烯哈密顿量与能带图(附Python代码)。
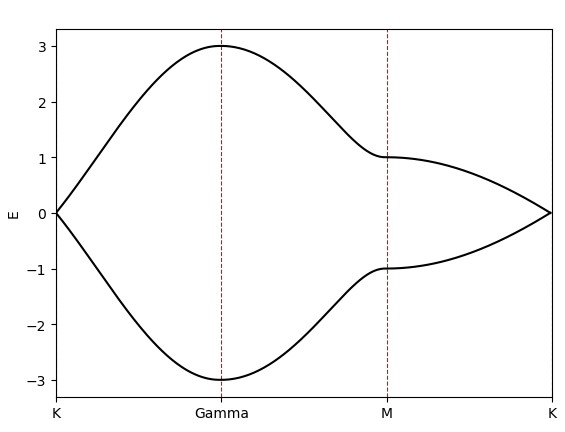
本篇画出石墨烯在各个高对称轴上的能带结构,有K-Gamma轴、Gamma-M轴、M-K轴。
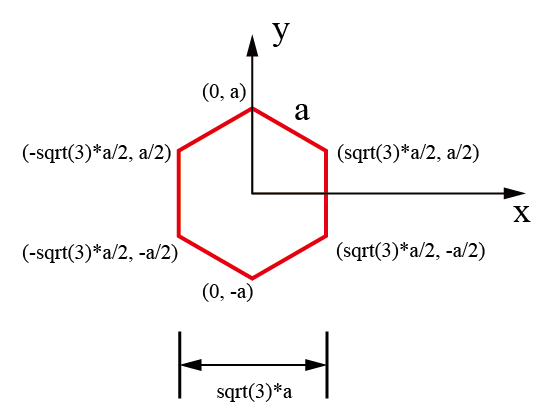
石墨烯在实空间中原子坐标示意图:
石墨烯在倒空间中高对称轴示意图:
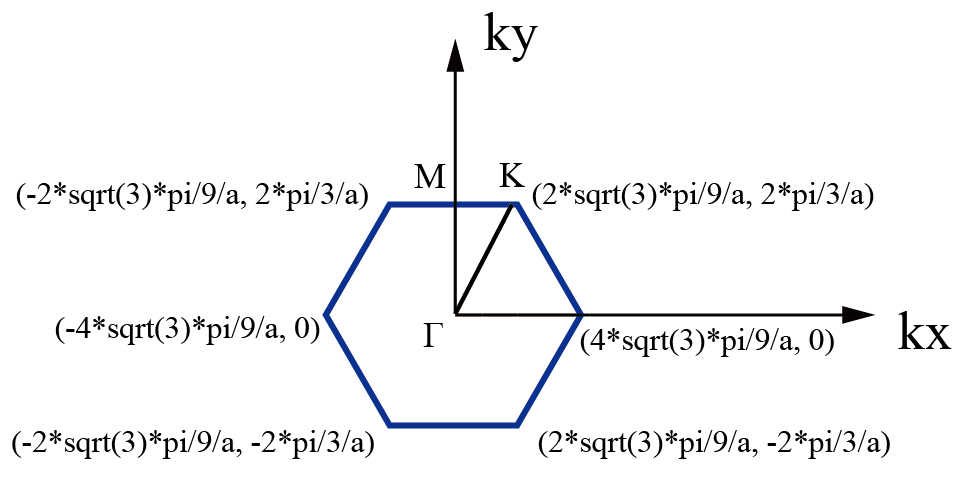
倒格子的坐标可参考:倒格子基矢的计算(附数值计算、符号计算Python代码)。
Python代码:
"""
This code is supported by the website: https://www.guanjihuan.com
The newest version of this code is on the web page: https://www.guanjihuan.com/archives/6260
"""
import numpy as np
import matplotlib.pyplot as plt
from math import * # 引入sqrt(), exp(), pi等
import cmath # 要处理复数情况,用到cmath.exp()
def hamiltonian(k1, k2, M=0, t1=1, a=1/sqrt(3)): # Haldane哈密顿量(a为原子间距,不赋值的话默认为1/sqrt(3))
h0 = np.zeros((2, 2), dtype=complex)
h1 = np.zeros((2, 2), dtype=complex)
h2 = np.zeros((2, 2), dtype=complex)
# 质量项(mass term), 用于打开带隙
h0[0, 0] = M
h0[1, 1] = -M
# 最近邻项
h1[1, 0] = t1*(cmath.exp(1j*k2*a)+cmath.exp(1j*sqrt(3)/2*k1*a-1j/2*k2*a)+cmath.exp(-1j*sqrt(3)/2*k1*a-1j/2*k2*a))
h1[0, 1] = h1[1, 0].conj()
# # 最近邻项也可写成这种形式
# h1[1, 0] = t1+t1*cmath.exp(1j*sqrt(3)/2*k1*a-1j*3/2*k2*a)+t1*cmath.exp(-1j*sqrt(3)/2*k1*a-1j*3/2*k2*a)
# h1[0, 1] = h1[1, 0].conj()
matrix = h0 + h1
return matrix
def main():
a = 1/sqrt(3)
Gamma0 = np.array([0, 0])
M0 = np.array([0, 2*pi/3/a])
K0 = np.array([2*np.sqrt(3)*pi/9/a, 2*pi/3/a])
kn = 100 # 每个区域的取点数
n = 3 # n个区域(K-Gamma,Gamma-M, M-K)
k1_array = np.zeros(kn*n)
k2_array = np.zeros(kn*n)
# K-Gamma轴
k1_array[0:kn] = np.linspace(0, K0[0], kn)[::-1] # [::-1]表示反转数组
k2_array[0:kn] = np.linspace(0, K0[1], kn)[::-1]
# Gamma-M轴
k1_array[kn:2*kn] = np.zeros(kn)
k2_array[kn:2*kn] = np.linspace(0, M0[1], kn)
# M-K轴
k1_array[2*kn:3*kn] = np.linspace(0, K0[0], kn)
k2_array[2*kn:3*kn] = np.ones(kn)*M0[1]
i0 = 0
dim = hamiltonian(0, 0).shape[0]
eigenvalue_k = np.zeros((kn*n, dim))
fig, ax = plt.subplots()
for kn0 in range(kn*n):
k1 = k1_array[kn0]
k2 = k2_array[kn0]
eigenvalue, eigenvector = np.linalg.eig(hamiltonian(k1, k2))
eigenvalue_k[i0, :] = np.sort(np.real(eigenvalue[:]))
i0 += 1
for dim0 in range(dim):
plt.plot(range(kn*n), eigenvalue_k[:, dim0], '-k')
plt.ylabel('E')
ax.set_xticks([0, kn, 2*kn, 3*kn])
ax.set_xticklabels(['K', 'Gamma', 'M', 'K'])
plt.xlim(0, n*kn)
plt.grid(axis='x',c='r',linestyle='--')
plt.show()
if __name__ == '__main__':
main()
计算结果为:
参考资料:
[1] 石墨烯的晶格和能带结构
【说明:本站主要是个人的一些笔记和代码分享,内容可能会不定期修改。为了使全网显示的始终是最新版本,这里的文章未经同意请勿转载。引用请注明出处:https://www.guanjihuan.com】
关老师好,实空间的原子坐标可以看懂,倒空间中高对称点的坐标是怎么得到的呢?始终困在原本的60或者120度夹角的基矢下。
参考:倒格子基矢的计算(附数值计算、符号计算Python代码)。
想请问一下,这个布里渊区是怎么算出来的,我算出的结果似乎有所不同
可以参考教材或这篇博文:倒格子基矢的计算(附数值计算、符号计算Python代码)。你说的结果不一样,有可能是因为a取的不同,这里a代表的是原子间距,而不是晶格常数,有一个根号3的关系。
您好,想请问一下,如果是三维的布里渊区路径要如何设置呢
如果是Gamma-X方向,ky=0,kz=0。如果是其他方向,比如kx=ky=kz方向,那么要共同变化取值,然后计算能带。
表面上看貌似没什么问题,但是如果将图片放大看,高对称点会有小问题。你在赋值的时候,比如设kn=5,k2_array[0:5] = np.linspace(0, M0[1], 5),前面指标对应的是0 1 2 3 4,所以高对称点对应的指标应该是4,不是5,即不是kn,而是kn-1,后面高对称点应该也是类似,我觉得这里要么就加上endpoint=false,要么采用arrage写法……
好像跟指标没关系吧,只要取点的个数是对应的就可以了。前面数组只是一个存放数据的变量而已。
高对称点指标肯定是有问题的 而且linspace也会重复取点,也就是说在中间的高对称点会取到两次
指标不影响结果呀,只是数据储存的中间过程。重复取点是有的,对画图也不影响。
好吧,你没懂我意思。简单来说,高对称点指标你的应该都差了1
嗯,是从0开始计,指标不影响结果。
石墨烯声子色散不应该是有6条分支吗
这个不是声子的,是电子的色散。
是我弄错了,谢谢
请问是否有声子色散的代码
这个我没考虑过。
最近邻那里的小括号是否有误,总觉得是t1*(k1*e1+k2*e2*k3*e3)
嗯,是这样的呀,刚好三项。
ax.set_xticks([0, kn, 2*kn, 3*kn, 4*kn])应该是ax.set_xticks([0, kn, 2*kn, 3*kn])
嗯,是的。代码已经修改了,不然运行会报错,感谢指出问题!